 mesitylene oxide
mesitylene oxide
Permanent Hair Removal 2: siRNA Boogaloo
This work began two years ago. You can see my first post on the topic here. I’ll give a summary and current status of the project, the rest of this post will cover technical details.
In short, the project went far but has not produced a successful treatment (as of 2024). I still believe that the procedure is technically sound and can be made to work, but at great cost and low (if not non-existent) commercial potential. siRNA is not cheap stuff even at scale, and has no chance of being approved anywhere for “aesthetic” procedures. The therapy has a number of not insermountable but certainly annoying intricacies - delivery and stability in ribonuclease rich environments aside, due to the nature of the hair follicle cycle and the RNAi process, treatment would likely have to be daily over an extended period (think 1-2 months)… at this point it is hardly preferable to electrolysis.
Perhaps more importantly, there are few things left here that still interest me - I wish to move on to other work. There are exciting things being worked on in the DIY biohacking and trans medicine communities… transfection of aromatase-regulating genes into testicular/ovarian tissue to produce natural levels of endogenous sex hormones, fluorescent skin markers allowing for facile monitoring of hormonal levels, a whole range of other gene therapies, womb transplants, et cetera. Perhaps I will attempt another such project one day, but for now my interests lie elsewhere.
Preliminary questions and reference literature
Non-specific advice from a nice Biotech Guy
with bulk RNA sequencing you can get intracellular siRNA's sequenced (maybe useful?)
shared the above site, extremely useful, shows lots of data on mrna and protein expression in different tissues (https://www.proteinatlas.org/ENSG00000168453-HR/tissue)
biotech statups doing therapeutics almost never succeed with the first trial, most are kept afloat by industry connections and rich guy money until they manage to get something to market, there was one really well connected guy who ran a biotech startup that took 20 years before they took something to market
filing a patent and leasing it (not selling usually!) to a larger pharma company is actually a very good way to get started with a biotech career! the people who succeed in biotech startups are usually older for the aforementioned reasons (although this is gradually starting to change, but still like 99% of people in successful biotech have phd's at least whereas in the tech sector you can just drop out of high school and if you are driven enough get funding from some venture capitalist lol)
patent that is abandoned cannot just be renewed, you need to add something novel (the patent office decides if you are novel enough) to the original patent in order to resubmit it
to get patents done by a good specialised law firm you'd pay 10k-50k$, to get it done on a budget you could maybe do it for 5k$. the actual patent application only costs like 20$ so if you get a bit of experience reading patents and put in some work and do a uni module on patent law you could easily apply for one yourself
RNAase contamination?
Useful anecdata was found on reddit1.
I did my PhD in an old school RNA biochemistry lab (though I didn't work on RNA). The folks in the lab who worked on RNA were vigilant about RNAses - they wore gloves, kept RNA on ice, washed all glassware in a mixture of alconox (sodium pyrophosphate + bac50) and KOH and baked glassware after washing in an ashing oven - but they scoffed at the sort of paranoia that most people have today. They never used RNAse Zap, or had special areas of the lab devoted to RNA work, or had special pipettes that were only used for RNA preps. Basically, most of the RNAses that will be in your sample are endogenous to your sample, unless you are a real slob. The key is to keep RNA on ice when it's not in e.g., phenol (this isn't required with Trizol or similar) so that RNAses cannot degrade an unprotected sample; work quickly; and use RNAse-free vessels (many plastic microcentrifuge tubes fit this requirement). Often they would do multiple rounds of phenol extractions, especially when moving from a large volume of lysate, like 50 ml, into smaller ones that fit into microcentrifuge tubes.A cheap decontamination solution was well investigated on the pipette jockey blog2.
My usual go-to for reverse engineering recipes are patents. You have to navigate some legalese but there’s definitely lot’s of good hints to be gleaned and even figures sometimes! Oh my! I searched pretty thoroughly for patents on RNase away and RNaseZap and came up empty handed. If they patented these solutions then the jig would be up, so I can understand why they didn’t. MSDSs are the next best source for a recipe, however something to keep in mind is that the laws vary by locality and time as to what the manufacturer HAS to disclose is in their product. For RNase Away several SDSs suggest that RNase away is nothing more than 0.5-1% NaOH and water. Due to it’s foamy nature though, I highly suspect that the other ingredient in RNase away is likely 5% SDS or sodium lauryl sulfate (SLS). So, SDS to denature proteins and breakup aggregates, and NaOH to hydrolyze proteins? Possible, but the NaOH concentration seems a bit low for that purpose, so if that is the recipe it likely doesn’t work great. RNaseZap MSDS revealed more recipe information. RNaseZap is composed of 1-5% SDS (5% probably), Sodium dichloroisocyanurate (0.5-1%) and (took a bit of digging) NaOH (?%, SDS states pH 7-8.5). Sodium dichloroisocyanurate is the interesting ingredient here, which in water will form hypochlorus acid (bleaching agent) that will destroy proteins, DNA and RNA.
I could’ve probably stopped there and been happy, but after a bit more digging found that Perkin Elmer manufactures AbSolve glassware cleaner/RNase decontamination reagent. No patent, but the SDS is very descriptive disclosing NaOH (2.5-10%), sodium hypochlorite (bleach, 2.5-10%), “alkylsulfonate steol” (<=2.5%), potassium citrate (2.5-10%) and 1-Ethyl-2-pyrrolidone (<=2.5%). The bleach, like before, destroys proteins and nucleic acids, the “steol” seems to be a trade name for SLS, and 1-Ethyl-2-pyrrolidone appears to help dissolve stuff…more better. I think the citrate could be used as a chelating agent to stabilize the bleach, or perhaps to balance pH.
Sometimes you strike reverse engineering gold. Qaigen filed a patent recently for “Permanent inactivation of nucleases “. Essentially their mixture is a simple solution of 5% SDS, 2% H2O2 and 6 mM EDTA, pH’d to 4.0 (pH’d with peracetic acid?). They likely imitated an older patent by the University of Montreal, “Synergistic detergent and disinfectant combinations for decontaminating biofilm-coated surfaces”, which used 1% EDTA, 5% H2O2, 1-2% SDS, 0.1% cetylpyridinium chloride and 1% peracetic acid. The Qaigen patent goes into great detail as to the rationale for the ingredients as well as some lovely gel images!
Decontamination Solution (v1.4)
10% Store bought bleach (2L per 20L)
1% NaOH (200g per 20L)
1% Sparkleen or similar powdered detergent (200g per 20L)
Instructions for use:
For most applications (Wiping down countertops, equipment, pipettes) the decon solution can be diluted 2-3X, wiped on, allowed to soak for several minutes and then rinsed off with distilled water and towels. More stubborn messes can be hit with undiluted mix. Glass and parts can be set to soak in straight or diluted (2-3X) decon mix, then washed as normal and rinsed with distilled water.
Don’t let the decon mix come in contact with anodized aluminum, it will take the color right off!How deep is hair below the skin?
“All of the machinery to make a hair lies below the scalp surface. Hairs typically extend 4-7 mm below the surface. It varies from patient to patient - and even varies to some degree for the same patient. The longest hairs on the scalp are the so called terminal hairs. The lie deep down in the fat layer.”3
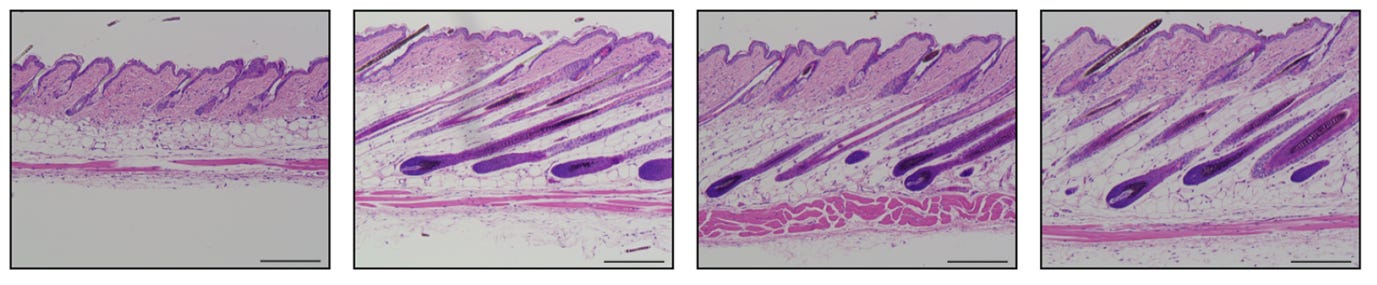
The scale bar in the image above is 200um, so yes the hair follicles in human skin are 1-5mm by depth. Own tests based on rough estimates of samples of ~5 hairs each:
arm hair: ~1.5-2mm
leg hair: ~2-2.5mm
tummy hair: ~2.5-3mm
siRNA stability / kinetics of siRNA degradation in vivo? Concentrations and off-target effects? does in vivo siRNA transfection require reagents?
Numerous papers/resources on stability456, concentration and off-target effects7891011 and in vivo transfection12 are available. Of note was that off-taget effects were more likely at higher concentrations (perhaps obvious), but of course at low concentrations the treatment is ineffective, highlighting the importance of dose ranging.
The issue then was that reported values spanned multiple orderds of magnitude. The genuine answer to all of the above is that it will depend on type of siRNA, which modifications have been made, what route of administration is used, and even the target gene and tissue.
Unmodified siRNA will be degraded very rapidly - given the time needed for transdermal delivery, this is not viable. Horizon was chosen as the siRNA provider. They were easy to work with and had good prices. A number of siRNA solutions were offered:
The OnTargetPlus siRNAs carry modifications on both strands in order to increase specificity and thereby significantly limit the number of off-targets. These siRNAs are not more resistant to RNases. The OnTarget Plus siRNAs are meant to be rapidly internalized by the cells, either by transfection or electroporation/nucleoporation methods. We do not have any document showing their degradation kinetics. We would not recommend OnTarget Plus siRNAs in nuclease-rich environments. Therefore, for in vivo applications, unless the siRNAs are protected by a carrier, we would not recommend OnTarget Plus siRNAs.
Please note that we propose two types of siRNAs which are more resistant to RNase degradation, the Accell siRNAs and the siSTABLE siRNAs, which are recommended for in vivo application. The Accell siRNA are extremely resistant self-delivering siRNAs. Accell siRNAs do not need any transfection reagent or electroporation. The Accell siRNAs are more recommended for local injections. You may find predesigned Accell siRNAs for your gene of interest.
The siSTABLE modification pattern dramatically enhances siRNA stability in nuclease-rich environments resulting in the siRNA reaching the target tissue to enable gene silencing. This modification is typically used for systemic injections into the blood stream, topical applications, and other applications where the siRNA is not protected by a carrier from nuclease degradation. We do not have pre-designed siSTABLE (except controls) in our catalog. You may convert any siRNA into siSTABLE siRNA with the "custom siRNA" page: https://horizondiscovery.com/en/ordering-and-calculation-tools/custom-sirna
The difference between Accell and siSTABLE is highlighted in these references from which you can also get delivery protocols:
https://horizondiscovery.com/-/media/Files/Horizon/resources/Reading-lists/sirna-in-vivo-ref-reading.pdf
In addition, for in vivo applications, we propose "in vivo" and "in vivo HPLC" processing options to help lower the toxicity to the animal. These options both include counter ion (Na+) exchange, desalting, sterile filtration, and endotoxin testing. Finally, the siRNA is lyophilized to allow greater solubility under the higher concentrations typically used for in vivo injections. The "in vivo" processing is highly recommended for any in vivo application.Given the above, Horizon Accell siRNA was chosen.
Accell siRNA is specially modified for use without a transfection reagent and works at a higher concentration than conventional siRNA with minimal disruption of the expression profile.
Annealed double-stranded RNA oligonucleotides
3'-UU overhangs
5'-Phosphate on antisense strand
Mass of each strand confirmed by MALDI-TOF mass spectrometry
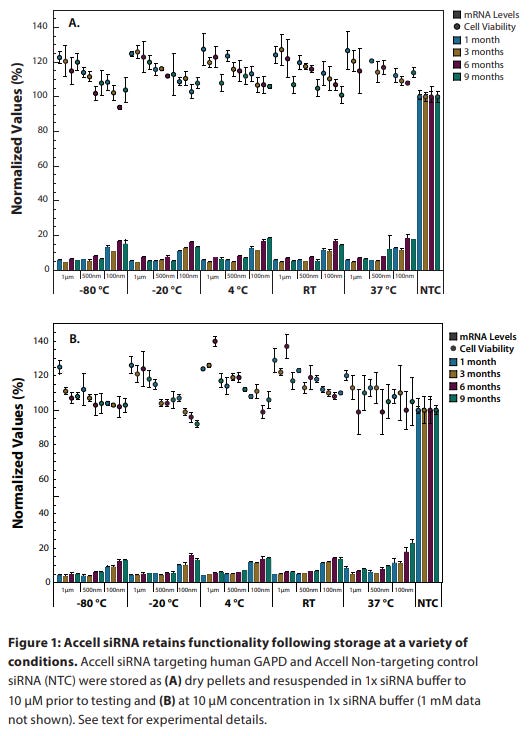
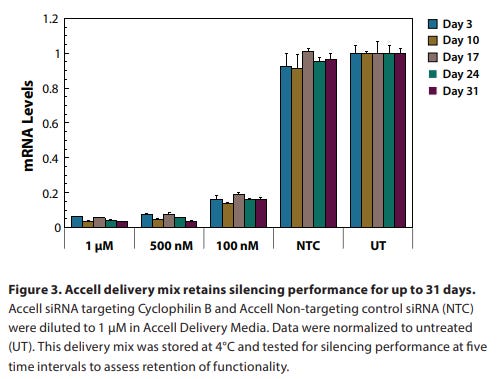
siRNA reagents are shipped as dry pellets at room temperature (23 °C). Under these conditions, they are stable for at least four weeks.
Upon receipt, siRNA reagents should be stored at –20 °C to –80 °C. Under these conditions, they are stable for at least one year.
siRNA should be resuspended in 1x siRNA buffer (Rnase-free, diluted from 5x siRNA buffer). RNase-free water (for short-term storage) is also appropriate for resuspension of concentrated stocks (20–100 µM). Alternatively, an RNase-free buffer (pH 7.3–7.6) may be used such as PBS.
Upon resuspension, aliquot the siRNA into small volumes and store at –20 °C to –80 °C. For best results, limit freeze-thawing of each tube to no more than five events. Under these conditions, the siRNA is stable for at least nine months.
Shelf stability from Horizon documentation is seen below.
Is the special accell delivery media needed?
While not required for use with Accell siRNA, this media provides appropriate serum-free conditions for Accell uptake while maintaining cell health. It may be replaced with another serum-free or low-serum media, or supplemented with additives known to be necessary for cell health. From an in-vitro transfection guide: “mix 7.5 μL of the 100 μM siRNA with 750 μL Accell Delivery Media. This is the delivery mix and can be used immediately.” TLDR only relevant for cell cultures.
Delivery case studies
Topical delivery of siRNA into skin using ionic liquids (10.1016/j.jconrel.2020.04.038.):
2.2. Synthesis of BDOA robed siRNA
siRNA robed with the ionic liquid (IL) benzyl dimethyl octyl ammonium (BDOA) were synthesized by acid-base neutralization as described previously [19]. Chloride salts were converted to hydroxide salts using Amberlite IRA-402 hydroxide anion exchange resin. BDOA salts were dissolved in ultrapure ddH2O at a concentration of 1.0% wt and mixed with excess resin for 1 h under constant agitation. Then, the solution was centrifuged to pellet the resin and collect supernatant. Complete anion exchange was verified by the lack of precipitate following dropwise addition of silver nitrate (2 g/mL in ddH2O). The final solution of BDOA was freeze-dried to remove ddH2O. Similarly, siRNA sodium salts were converted to acidic form using Amberlite IR-120 hydrogen cation exchange resin. Complete cation exchange was verified by titration. The final solution of hydrogen form of siRNA was freeze dried to remove ddH2O. Acidic groups on siRNA were neutralized by addition of equivalent BDOA hydroxide in methanol. Neutralization was made overnight at room temperature with constant agitation. Methanol was then removed by rotary evaporation and subsequent freeze-drying, and BDOA robed-siRNA was stored at −20 °C until further use.
2.3. Synthesis of CAGE and characterization
CAGE 1:2 was prepared as previously reported [20]. Two equivalents of neat geranic acid (20 g, 0.119 mol), recrystallized five times at −70 °C in acetone, were added to one equivalent of choline bicarbonate (80 wt% solution, 12.274 g, 0.059 mol). The mixture was stirred at 40 °C until CO2 production ceased. The rotary evaporation was used at 60 °C for 20 min to remove the remaining water, and further dried in a vacuum oven for 48 h at 60 °C. CAGE 1:2 was characterized as previously described and reported [20].
2.9. In-vivo siRNA-IL studies
The experiments were performed using female SKH-1E hairless mice of 6–8 weeks. The animals were kept in a controlled temperature (24 to 26 °C), a daily 12:12 h light/dark cycle and food and water ad libitum. The protocol was approved by the Harvard University Animal Committee. All IL formulations (CAGE, CAGE-siRNA, BDOA, BDOAsiRNA and CAGE-BDOA-siRNA) were tested with the GAPDH siRNA (50 μM = 665 ng/uL = 0.665 ug/uL) in healthy animals to verify the knockdown of the GAPDH protein. The mice were treated for 4 consecutive days, once a day with a volume of 25 μL of each formulation. On the fifth day, the animals were euthanized, the treated skin area was harvested for further analysis. 25% CAGE 1:2 was utilized in the siRNA treatment groups. In vitro studies were performed using porcine skin, which poses a significantly tougher barrier compared to mouse skin used for in vivo experiments. Hence, 25%v/v CAGE was used for in vivo studies compared to 50%v/v used for in vitro studies.Transdermal delivery of Fn14 siRNA using a novel composite ionic liquid for treatment of psoriasis-like skin lesions (10.1016/j.jconrel.2023.12.009):
Transdermal delivery of siRNAs for Fn14 inhibition is challenging. In this study, we developed a composite ionic liquid (CIL) for the transdermal delivery of Fn14 siRNA (siFn14) into keratinocytes, with the aim of modulating the inflammatory response associated with psoriasis. The results showed that CIL-siFn14 effectively suppressed Fn14 expression, resulting in a reduction in both the Psoriasis Area and Severity Index (PASI) score and skin thickness.
2.1. Preparation and characterisation of CIL
The ILs were synthesized through an acid-base neutralisation reaction by mixing choline bicarbonate (Ch) with geranic acid (Ger) at a ratio of 2:3 and Ch with 3-phenylpropionic acid at a ratio of 1:2. The specific procedure was as follows: 10 g (0.0666 mol) of 3-phenylpropionic acid was dissolved in 60 mL of anhydrous ethanol, and 5.425 mL (0.0333 mol) of an 80 wt% aqueous solution of Ch was added dropwise. The mixture was stirred at room temperature for 12 h until CO2 was no longer produced. Ethanol and H2O were removed using a rotary evaporator (60◦ C for 6 h), and then the mixture was further dried in a vacuum freeze dryer (AST-V8-2MJ) for 48 h to obtain 3-phenylpropionic acid hydrocholine carbonate ([Ch][Hyd] 2). Next, 3.288 g (0.0193 mol) of Ger was dissolved in 60 mL of anhydrous ethanol, and 4.73 mL (0.0289 mol) of an 80 wt% aqueous alkali solution was slowly added dropwise. The mixture was stirred at room temperature for 12 h until CO2 was no longer produced. Ethanol and H2O were removed using a rotary evaporator (60◦ C for 6 h), and subsequently, the mixture was further dried in a vacuum oven (60◦ C for 48 h) to obtain [Ch]2[Ger]3. Then, [Ch][Hyd]2 and [Ch]2[Ger]3 were mixed in a 1:1 volume ratio using a rotary stirrer (EYELA N-100) for 2 h to obtain the dried CIL. The ILs were characterised using Fourier-transform infrared (FT-IR) spectroscopy, and nuclear magnetic resonance (NMR). FT-IR spectra were collected on a NicoletTM iSTM 5 FT-IR spectrometer (Thermo Fisher Scientific, Madison, Wisconsin, United States of America). 1H NMR spectroscopy was performed using a Bruker Avance III 400/600 MHz spectrometer (Bruker, Bern, Switzerland), and the ILs were NMR analysed using dimethylsulfoxide (DMSO)-d6. Lastly, the conductivity of the aqueous diluted ILs was determined using a conductivity meter (INESA DDS-307 A) under stirring conditions at 25◦ C. The measurements were repeated thrice for each sample and averaged.
2.2. Preparation and characterisation of CIL-siFn14 formulation
To prepare the CIL-siFn14 formulation, 1 OD of siFn14 was diluted with 250 μL of DEPC, and then 750 μL of CIL (siFn14:CIL = 3:1) was added to achieve a concentration of 125 nM of CIL-siFn14. The homogeneous CIL-siFn14 was achieved by rotating it for 24 h using a rotary stirrer. The CIL was analysed by incubating HaCaTs with 10 μM siFn14 at a volume ratio of 1:50 for 30 min at 25◦C. Subsequently, the solution was dialysed in a beaker using a dialysis bag (10,000 molecular weight cutoff, Invitrogen, Waltham, MA, USA) with phosphate-buffered saline (PBS) for 24 h under continuous stirring to eliminate any interference from the IL in subsequent steps. Circular dichroism (CD) spectra of Naked-siFn14 and CIL-siFn14 were recorded from 200 nm to 310 nm using a CD spectrometer equipped with a fluorescence photomultiplier tube (Aviv Biomedical, Inc., Lakewood, NJ, USA) at 24.98◦C, and the data were averaged from three independent measurements.
2.8. Mouse model and treatment
Wistar rats were randomly divided into four groups (n = 3/group): PBS, 30% CIL, 60% CIL, and 100% CIL. BALB/c mice were randomly divided into five groups (n = 5/group): control (PBS), imiquimod (IMQ) model, IMQ + DMSO-siFn14, IMQ + Naked-siFn14, and IMQ + CIL- siFn14. siFn14 was used at a concentration of 125 nM. Except for the control group, mivamotte cream was applied to the backs of the mice using a glass rod at a dose of 62.5 mg once daily for 7 d. Twelve hours after mivamotte cream application, 100 μL of the different formulations of the above drugs were applied to the same area. A second set of BALB/c mice with the same acclimatisation period and skin preparation were randomly divided into two groups for biosafety evaluation (n = 3/group): blank control (300 μL DEPC water) and CIL-siFn14 (300 μL IL-siFn14). The backs of the mice were coated in the treatment each day for eight consecutive days. On the ninth day, the mice were euthanised, and their back skin was collected.Transdermal Delivery of siRNA through Microneedle Array (10.1038/srep21422):
Microneedles.
Figure 1 present the SEM image of a typical silicon microneedle array patch, which indicates that the microneedles have uniform morphology and geometry. They exhibit pyramidal shape and the radius of tip is below 1 μm. The length of these fabricated needles is 200± 7 μm (n= 900needles/array and 121 arrays per wafer). The spacing between microneedles is 90μm with slight variation (i.e. less than 1μm). For each single microneedle, the surface is rough and layer upon layer.
Silencing of Gapdh gene expression.
Mice were terminated at 24 h after the treatment, and the ear skin was excised for RNA extraction. Gapdh mRNA levels in the epidermis were measured (Fig. 4). A marked dose-dependent reduction of Gapdh gene expression was detected in the skin treated with the siRNA (5, 10 or 15 μg/μl, 15 ug/uL = 15000ng/nL = 1130 uM) compared with the contralateral flank skin without treatment. The inhibition percentages are around 15%, 40% and 66%, respectively. The expression of Gapdh was normalized by the expression of m-18s.CAGE and water?
Computational and experimental characterization, namely viscosity, conductivity, and self-diffusion coefficient, were employed here to understand the properties of equimolar CAGE (1:1 choline: geranic acid) in the presence of varying amounts of water. It was found that under stored conditions, 1:1 CAGE contained up to 0.20 mole fraction water. Experimental and computational studies indicate that microscopic intra-ionic interactions within CAGE are not substantially changed until the water content exceeds 0.65 mole fraction. At this point, we theorize that the geranate ions undergo reorganization to minimize contact between the hydrophobic tails and the water molecules. This is evidenced by the plateau in viscosity at this mole fraction, and the increased interactions between the tails of the anions. This suggests that CAGE could be used without pre-drying in most applications and can be diluted to induce the organization of the anions where desired.
(https://www.researchgate.net/publication/333498844_The_Influence_of_Water_on_Choline-Based_Ionic_Liquids)CAGE preparation
CAGE ionic liquids were prepared as described previously.3 Briefly, commercial geranic acid (85%, stabilized, Sigma Aldrich, St. Louis, MO, USA) was recrystallized in acetone at −80°C (70% [wt] geranic acid solution) 5–7 times. Purity was confirmed with 1H-NMR prior to preparing mixtures. Next, choline bicarbonate (80% in water, Sigma Aldrich) was mixed with pure geranic acid at a choline:geranic acid molar ratio of 1:1, 1:2, 1:3, and 1:4. The mixture was rotary-evaporated at 1 atm and 40°C until carbon dioxide production ceased. The mixture was then rotary evaporated at 20 mBar and 60°C for a minimum of 2 h (until water evaporation ceased). The mixtures were then assessed with 1H-NMR to confirm that stoichiometry was maintained during preparation. The integral value for the methyl peak from choline was set to 9, then the integral values for both lone hydrogen atoms on geranic acid's alkene carbons was compared to expected values (expected integral values were 1, 2, 3, and 4 for the 1:1, 1:2, 1:3, and 1:4 molar ratios, respectively).
(Choline and geranic acid (CAGE) ionic liquids inhibit both elastase activity and growth of oral bacteria https://onlinelibrary.wiley.com/doi/full/10.1002/jbm.a.37485)More Papers (mostly from gonzalez-gonzalez)
10.1038/jid.2010.426 Use of Self-Delivery siRNAs to Inhibit Gene Expression in an Organotypic Pachyonychia Congenita Model
Accell, in-vitro epidermis model
Two of the major obstacles to siRNA delivery to skin are penetration through the stratum corneum barrier and efficient functional siRNA uptake by keratinocytes, including incorporation into the RNA-induced silencing complex. The stratum corneum barrier, composed of terminally differentiated squamous cells containing a hydrophobic intercellular lipid matrix, prevents efficient penetration of large (4500 Da), charged molecules (e.g., siRNA is highly anionic and B13,000 Da) that are topically administered. The advantages and disadvantages of a number of methods to facilitate stratum corneum penetration have been summarized in a recent review (Geusens et al., 2009), including tape stripping, ballistic delivery, microneedle application, intradermal injection, sonophoresis, electroporation, jet injection, iontophoresis, chemical enhancers, lipid-based systems, and chemical depilation.
In this study, we have evaluated Accell self-delivery siRNAs (CBL3 and K6a_513a.12) and demonstrate specific target inhibition in epidermal skin equivalents expressing reporter proteins or skin equivalents prepared from PC patient-derived keratinocytes by repeated and non-invasive imaging over the course of the experiment. Recently, self-delivery siRNAs delivered by microneedle arrays to transgenic reporter mice have been shown to silence target gene expression (Gonzalez-Gonzalez et al., 2010b), suggesting that this approach may have clinical utility, in which direct, local administration is feasible. Owing to the proprietary nature of the self-delivery siRNAs (at least for the Dharmacon and RXi inhibitors), it is unknown if these inhibitors are working through a similar mechanism(s) such as conjugations of lipids or steroid moieties, as demonstrated by Alnylam Pharmaceuticals or through a distinct mechanism.
The availability of self-delivery siRNAs that are readily taken up by skin keratinocytes, coupled with a stratum corneum penetration strategy (e.g., microneedles or topical formulations), may result in patient-friendly delivery of sufficient quantities of inhibitor to result in clinical efficacy.
siRNA was added to a final concentration of 2 uM (optimal concentration determined by dose–response studies in primary human keratinocytes, data not shown; this concentration is also consistent with the manufacture’s recommendations) directly to the medium in the lower chamber, leaving the surface of the epidermal equivalent dry.10.1016/j.jconrel.2012.12.030 Gene silencing following siRNA delivery to skin via coated steel microneedles
Accell, microneedles, in-vivo
Self-delivery siRNA targeting the reporter (luciferase/GFP) gene was coated onto microneedles and delivered to mouse footpad. Quantification of reporter mRNA and intravital imaging of reporter expression in the outer skin layers confirmed functional in vivo gene silencing following microneedle delivery of siRNA. The use of coated metal microneedles represents a new, simple, minimally-invasive, patient-friendly and potentially self-administrable method for the delivery of therapeutic nucleic acids to the skin.
Two μL of siRNA coating solution (70 mg mL−1 for Accell sd-CBL3 and 80 mg mL−1 for Accell sd-Control) in PBS was loaded into a pipette tip reservoir for coating. The steel microneedle devices (4 devices per treatment group; each with 10 microneedles per array) were coated with siRNA using the coating method described in Fig. 1A to provide a theoretical loading of 35 μg and 40 μg Accell CBL3 and Accell control sd-siRNA coated onto each microneedle device, respectively. Coated microneedles were maintained at 4 °C for up to 18 h before use.10.1038/mt.2010.126 Silencing of reporter gene expression in skin using siRNAs and expression of plasmid DNA delivered by a soluble protrusion array device (PAD)
Accell, microneedles, in-vivo
Despite rapid progress in the development of potent and selective small interfering RNA (siRNA) agents for skin disorders, translation to the clinic has been hampered by the lack of effective, patient-friendly delivery technologies. The stratum corneum poses a formidable barrier to efficient delivery of large and/or charged macromolecules including siRNAs. Intradermal siRNA injection results in effective knockdown of targeted gene expression but is painful and the effects are localized to the injection site. The use of microneedle arrays represents a less painful delivery method and may have utility for the delivery of nucleic acids, including siRNAs. For this purpose, we developed a loadable, dissolvable protrusion array device (PAD) that allows skin barrier penetration.10.1007/978-1-4939-3148-4_1 Imaging Functional Nucleic Acid Delivery to Skin
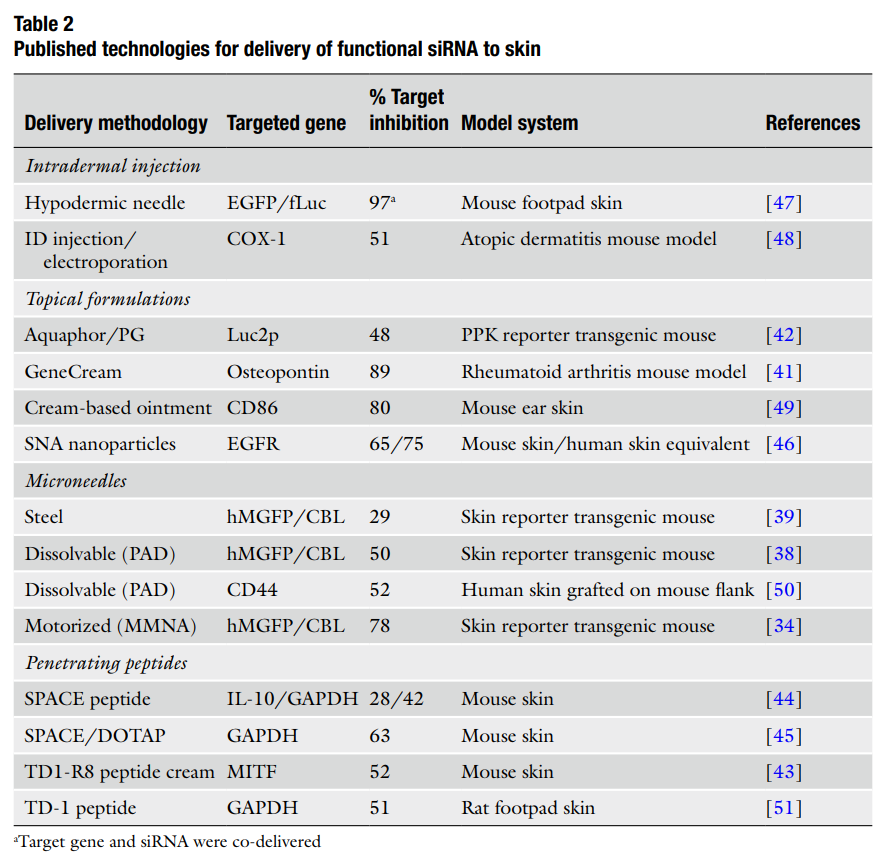
Method and Results
Genome screening
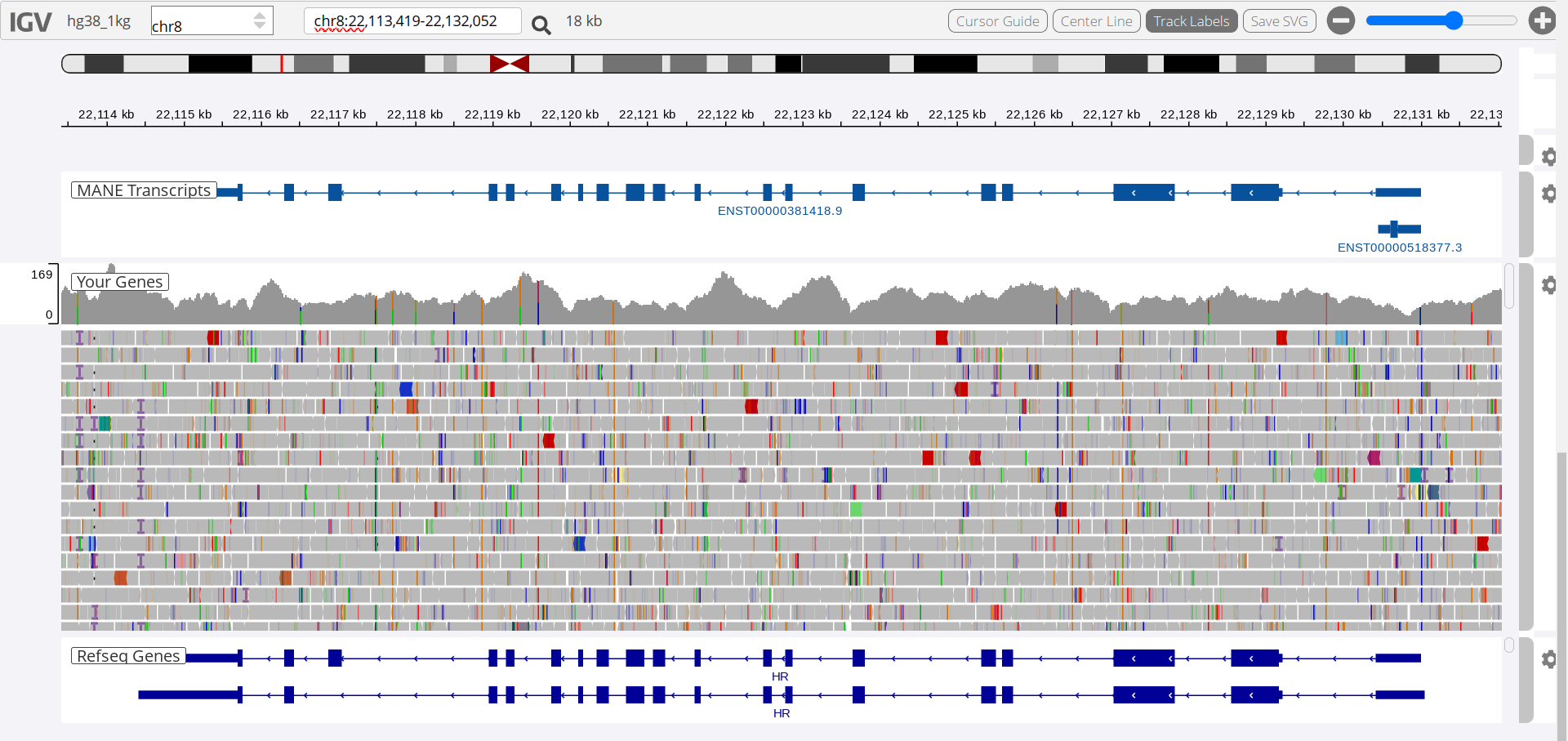
siRNA binds selectively to mRNA sequences (usually 21bp) - even one bp mismatch will significantly lower binding affinity. Having found a commercial supplier of siRNA targetting the HR gene I needed to check for any discrepancies between the reference human genome and my own.
I had myself screened by Nebula Genomics, 249$ for 30x sequencing with good online genomics tools and processing of all your data, a bargain. I had a number of SNPs in my HR gene but only one of these was located at an exon. I was able to confirm that none of the siRNAs would be affected by this SNP.
I also had a full genome and cellular mRNA sequence done by BGI. Their customer service was great, a sales rep took me out for coffee and I got souvenirs!

That being said, unlike Nebula I had to ship them samples on dry ice (necessary for mRNA), they throw the raw unprocessed data back at you and sadly the results weren’t as good (for a variety of reasons… this was around Chinese New Year too so there was some delay with the screening, but overally they did everything significantly faster and more reliably than BGI). Also unless you are sending samples in bulk they are more expensive. For complex screening work I’d go with BGI but in this case honestly the Nebula screen was much better value for money.

Reagents and Equipment
Chemicals for CAGE were acquired from LEAP CHEM Co., Ltd. They were good to do business with: fast reply times, good prices and efficient customs clearance.

Blunt 34G needles were acquired from CelLink


A variety of microneedle devices were procured via amazon.

Human HR Accell siRNA SMARTPool was acquired from horizon13. The simple microneedle rollers were found to be easiest to use.
Experimental
Ionic Liquid
Small scale CAGE 1:2 hotplate synthesis:
geranic acid (5 g, 0.119/4 mol) was added to choline bicarbonate (80 wt% solution, 3.0685g, 0.059/4 mol), mixture was heated at 50C until CO2 production ceased. No further changes were observed until a temperature of 100C, whereupon light bubbling occurred. The mixture was allowed to cool to room temperature.
Possible ionic liquids:
choline hexanoate
choline urea
choline geranic acid
siRNA
Administration:
wax hair
apply warm compress / steam / shower
apply [some concentration] (0.5-5000uM?) of [ionic liquid/siRNA solution]
[optional microneedling step]
[intradermal injection?]
siRNA resuspension
50nmol of siRNA was added to 1mL 1x buffer (made with 200uL 5x buffer, 800uL saline) and, once dissolved, filtered (skip filtering to avoid liquid loss) added to a sterile vial to produce a solution of 50uM concentration.
First Tretment
Geranic acid (2.5 g, 0.119/4 mol) was added to choline bicarbonate (80 wt% solution, 1.53425g, 0.059/4 mol) to produce ~3g IL. 0.5g ionic liquid was poured into a presterilised vial and 5nmol siRNA (N.B. ONTarget siRNA was used for this trial which likely degraded fast, never even having a chance at penetrating the skin) was added (0.1mL of 25uM) to produce a solution of concentration around 5uM. This mixture was poured over a waxed arm that had been soaked in hot water. After a few minutes clingfilm was applied.
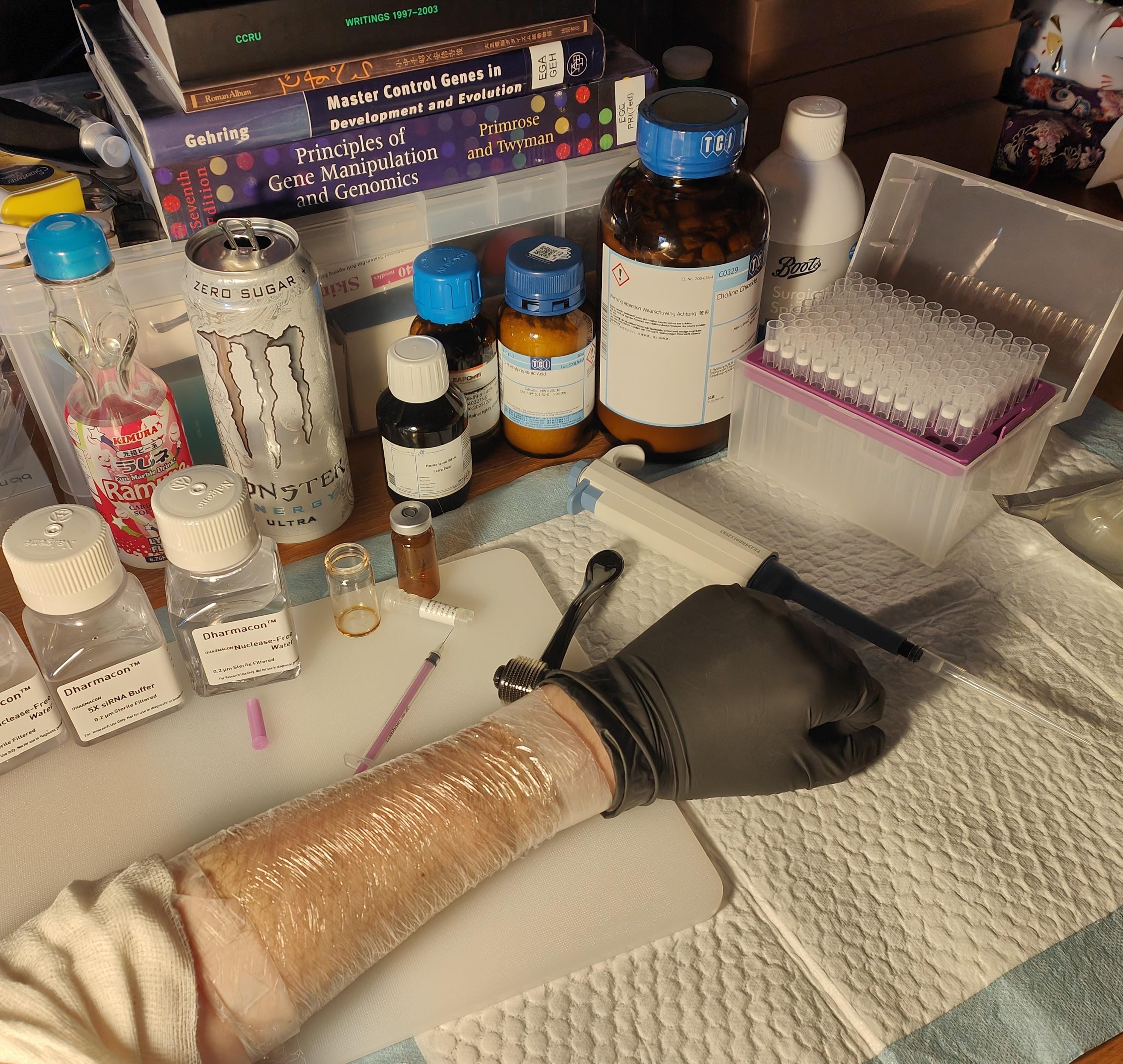
No negative side reactions were observed over the upcoming weeks. No pain was observed. The procedure was repeated, this time using microneedle rollers - pain subsided after a few hours. An eczema-like reaction was observed over the the upcming days. Hair proceeded to grow back as normal.
Subsequent treatment (few months later)
N.B. You actually need a pretty minimal setup. Remember, once applied the siRNA is in a ribonuclease-rich environment anyway. No need to sterilise your working surface (beyond basic cleaning) etc. You need a respirator, gloves, sterile syringes, siRNA vials (resuspension can be performed *in* the vial that the siRNA came in, they hold about 2mL), and any further reagents/equipment used for transdermal delivery (e.g. microneedle roller). Each time you wish to carry out the procedure, take out a fresh aliquot of resuspended siRNA with a sterile syringe and use as needed.
As described, 50nmol Accell siRNA was resuspended in 1x siRNA buffer, kept at -20C in freezer between use.
Day 0: after waxing, 0.2mL 50uM solution applied directly to skin using microneedle roller and small amount of E45 cream.
Day 4: no bad reaction from first treatment observed. Hairs on test arm seem easier to pluck (possibly due to microneedling)? 0.2mL 50uM applied directly using microneedle roller (no E45).
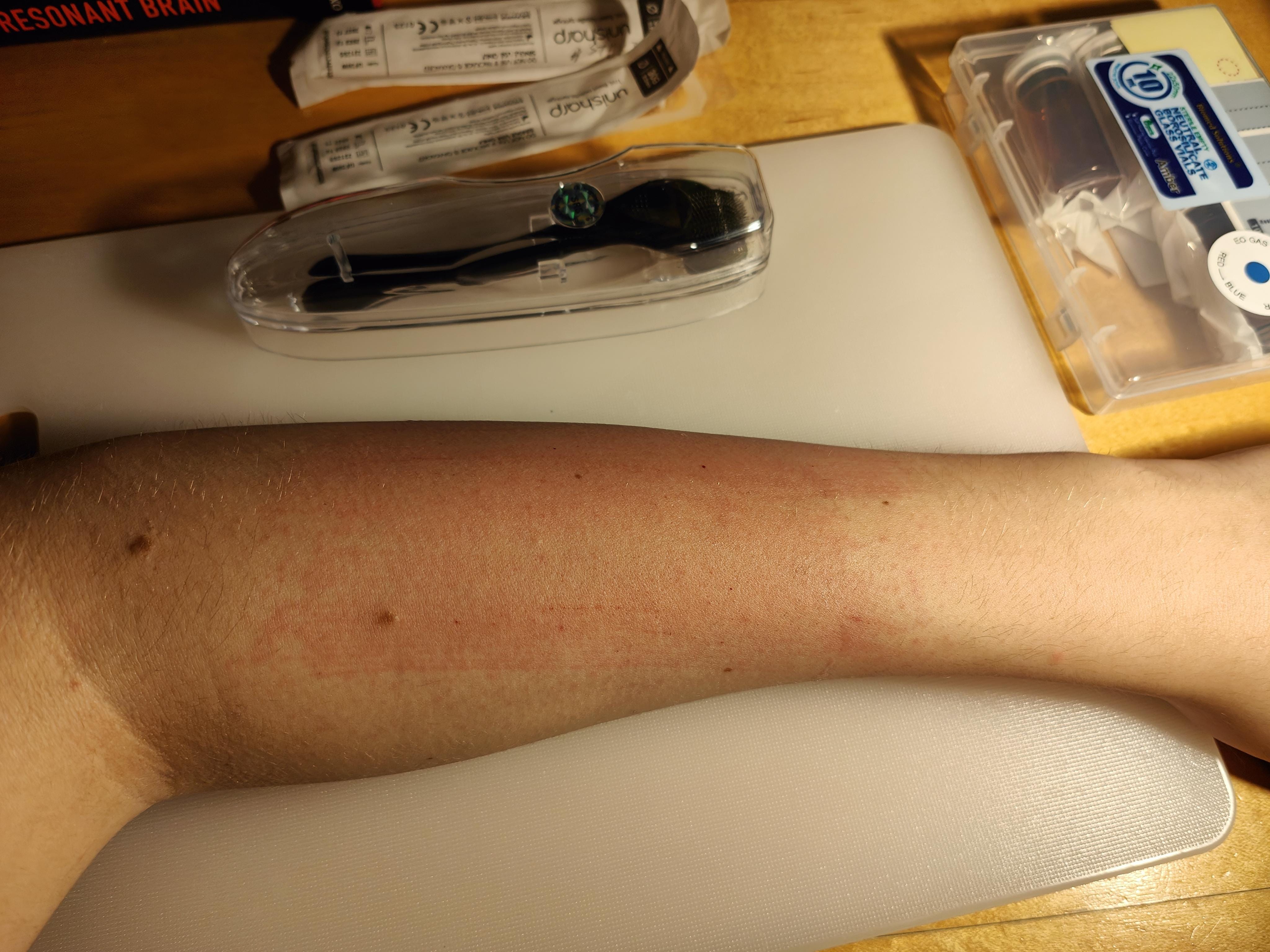
Hair proceeded to grow back as normal.
Further Investigation, to-do
Continue the treatment daily over an extended period e.g. a month
Use the 34G needles to deliver siRNA into hair follicle
Try using ionic liquid with Accell siRNA
Try intradermal injection?
???
This project has cost about 8,000$ in total so far (funding I managed to secure from a kind donor), not something I could ever afford myself. If you enjoy this work, do consider donating.
Notes
Influence of diverse storage conditions of double-stranded RNA in vitro on the RNA interference efficiency in vivo insect Tribolium castaneum. https://pubmed.ncbi.nlm.nih.gov/36086883/
Overcoming Barriers for siRNA Therapeutics: From Bench to Bedside https://pmc.ncbi.nlm.nih.gov/articles/PMC7600125/
siRNA Off-Target Effects Can Be Reduced at Concentrations That Match Their Individual Potency https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0021503
Off-target effects of RNAi correlate with the mismatch rate between dsRNA and non-target mRNA https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8583100/
A computational study of off-target effects of RNA interference https://pubmed.ncbi.nlm.nih.gov/15800213/